Methylation is a wide-spread and essential post-translational modification. Protein methyltransferase transfers the activated methyl group from S-adenosyl-L-methionine (SAM) to amino acid residues on a nitrogen, oxygen or sulfur center. Earlier studies identified only a few targets or methylation on limited amino acid residues. Therefore, it remains an intimidating challenge for global identification of methylated proteins.
The research work led by Prof. ZHAO Zongbao and Prof. ZOU Hanfa from Dalian Institute of Chemical Physics, Chinese Academy of Sciences, described major progress on proteomic analysis of protein methylation.
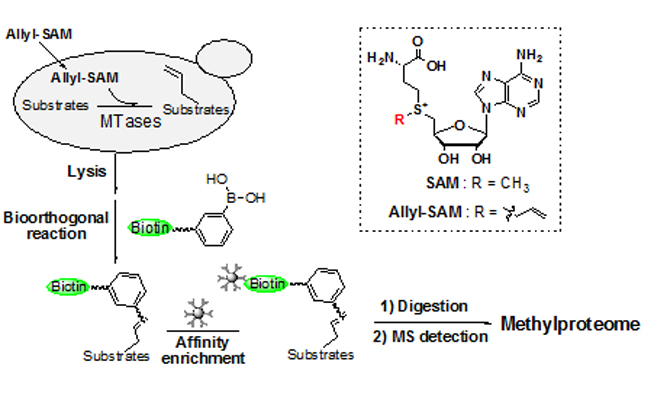
DICP Researchers Devised a Chemical Proteomic Approach to Capture Methylated Protein (Image by NING Siyang)
Researchers devised a chemical proteomic approach to capture methylated protein. This strategy involved in vivo protein allylation, chemical tagging, affinity enrichment, and target identification based on tandem mass spectrometry analysis. The study identified 167 hits, covering many proteins with known methylation events on different types of amino acid residues.
Moreover, researchers confirmed the presence of methylation events on multiple residues in two hits, namely, hexokinase isoenzyme 2 and 6-phosphogluconate dehydrogenase. Both of them are the key enzymes in the central carbon metabolism. The results provided comprehensive database that may inspire further studies on the biological function of protein methylation.
The work was published on Chemical Communications (2016, 52, 6689). (Text and Image by NING Siyang)






